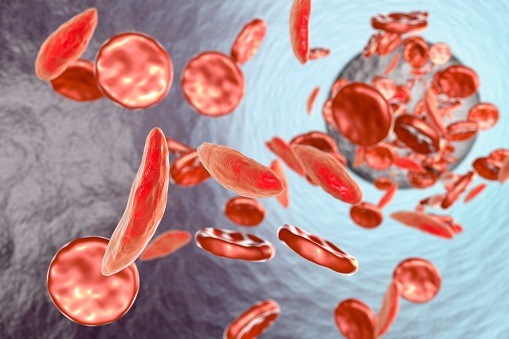
Sickle cell disease refers to a group of inherited red blood cell disorders. Anywhere between 70,000 and 80,000 Americans have sickle cell disease. One of the most common types of sickle cell disease is sickle cell anemia, also referred to as hemoglobin SS or HbSS. In sickle cell anemia, a person inherits two hemoglobin S genes from their parents. Sickle cell anemia, in addition to being one of the most common, is also considered the most severe form of sickle cell disease.
In healthy patients, red blood cells have a round shape and can travel through the blood vessels with ease. However, sickle cell anemia patients have sticky, rigid red blood cells that lack flexibility. They have a banana or “sickle” shape and are prone to becoming trapped in the blood vessels, leading to strained or blocked blood and oxygen flow.
Who Is At Risk?
Sickle cell disease is an inherited disorder. A baby must inherit the sickle cell gene from both of their parents. It is also possible to inherit the defective gene from only one parent—leaving a person with sickle cell trait. These people are usually healthy but risk passing the gene on if they ever have children of their own.
Signs of sickle cell disease usually begin as early as age five months. In the United States, all newborns are tested for sickle cell disease, so treatment can begin early.
Signs and Complications
Symptoms may change overtime and vary from person to person. Some symptoms of sickle cell anemia include:
- Anemia—a condition where patients have a low red blood cell count. Sickle cells are more prone to breakage and dying at a faster rate than the body can replenish them. This in turn means that oxygen cannot flow through the body quickly enough, causing fatigue.
- Pain episodes, referred to as crises, are a result of blocked blood flow when the sickle cells cannot pass through the blood vessels. Duration and intensity of pain episodes vary from patient to patient—they may last just a few hours or several weeks, and may occur infrequently or several times a year. Patients with bone and joint damage may also experience chronic pain in the bones.
- Swelling may occur of restricted or blocked blood flow.
- Frequent infections. Sickle cells may cause damage to the spleen, leaving the body less equipped to fight off infection. Because of this, doctors may recommend vaccinations to prevent illnesses such as pneumonia that could be fatal in immune-compromised patients.
- Delayed growth. Since red blood cells are responsible for the oxygen and nutrients vital for growth, the shortage in patients with sickle cell anemia may lead to slowed growth in babies and delayed puberty in adolescents.
- Vision problems may manifest if the blood vessels of the eyes contain sickle cells, leading to retina damage.
Sickle cell disease is linked to many possible complications, including:
- Acute chest syndrome, which could be fatal, may be the result of a lung infection or could occur when sickle cells block the lung’s blood vessels. Signs include chest pain, fever, and trouble breathing.
- Acute pain crisis, or sickle cell or vaso-occlusive crisis. This may come on suddenly when oxygen flow drops due to blocked blood vessels. It’s not clear what triggers the pain, which is commonly reported in the abdomen, low back, chest, arms, and legs.
- Brain complications, which could include clinical or silent stroke. Patients with sickle cell disease may exhibit signs of stroke, but adults with sickle cell anemia may suffer from a silent stroke, which can be detected through an MRI. This brain injury can inhibit daily functions and may cause problems with learning and decision-making.
- Cardiac complications. Patients who have underwent many blood transfusions may sustain heart damage caused by iron overload. Other complications may include ischemic heart disease and pulmonary hypertension.
- Pregnancy complications. Expecting mothers with sickle cell disease may have increased blood pressure and blood clots. There is also a higher risk for miscarriage, premature birth, and low birth weight.
Other possible complications include kidney problems, liver complications, priapism, and ulcers in the leg.
Treatment Options
The only way to cure sickle cell disease is by bone marrow or stem cell transplantation. Donors are often a close, immediate familial match. Both options are high-risk surgeries with significant possible adverse outcomes. For this reason, surgical treatment is usually only performed in patients with severe disease.
Other treatments target complications associated with the disease. For instance, patients may take two daily doses of penicillin to reduce the risk of contracting certain infection. Some patients stop this treatment at age five, but patients with sickle cell anemia may choose to continue taking it for life.
Another option is hydroxyurea, which increases fetal hemoglobin in the blood and reduces some of the complication’s side effects, including pain and anemia. Its long-term effects have yet to be established, and it has been rarely associated with worsening anemia.
Some patients may undergo blood transfusions. An acute transfusion may be performed to treat severe anemia, if a patient has suffered a stroke, or in the event of acute chest crises or organ failure. Red blood cell transfusions are also performed, to increase red blood cell count. Patients who have had an acute stroke may require regular or ongoing blood transfusions to help prevent another stroke. This may be recommended in patients to prevent a first stroke as well, and in patients whose complications do not improve with hydroxyurea.
Sources: Mayo Clinic; National Heart, Lung, and Blood Institute; MedlinePlus; Centers for Disease Control and Prevention; Sickle Cell Anemia News




 © 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.
© 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.